



AlphaFold
本软件包提供了AlphaFold v2推理流程的实现。为简单起见,在本文档的其余部分中我们将该模型称为AlphaFold。
我们还提供:
-
AlphaFold-Multimer的实现。这是一个正在进行的工作,AlphaFold-Multimer预计不会像我们的单体AlphaFold系统那样稳定。阅读指南了解如何升级和更新代码。
-
包含更新后的AlphaFold v2.3.0模型和推理程序的技术说明。
-
CASP15基线预测集以及对所进行的任何手动干预的文档。
任何使用此源代码或模型参数而产生研究结果的出版物都应引用AlphaFold论文,如适用,还应引用AlphaFold-Multimer论文。
请同时参考补充信息以获取该方法的详细描述。
您可以使用此Colab笔记本中略微简化的AlphaFold版本或社区支持的版本(见下文)。
如有任何问题,请通过alphafold@deepmind.com联系AlphaFold团队。
安装和运行您的第一个预测
您需要一台运行Linux的机器,AlphaFold不支持其他操作系统。完整安装需要高达3 TB的磁盘空间来�存储遗传数据库(建议使用SSD存储),以及现代NVIDIA GPU(内存更大的GPU可以预测更大的蛋白质结构)。
请按以下步骤操作:
-
安装Docker。
- 安装NVIDIA Container Toolkit以支持GPU。
- 设置以非root用户身份运行Docker。
-
克隆此仓库并进入其目录。
git clone https://github.com/deepmind/alphafold.git cd ./alphafold -
下载遗传数据库和模型参数:
-
安装
aria2c。在大多数Linux发行版中,它可通过包管理器以aria2包的形式获得(在基于Debian的发行版中,可以通过运行sudo apt install aria2来安装)。 -
请使用脚本
scripts/download_all_data.sh下载并设置完整数据库。这可能需要相当长的时间(下载大小为556 GB),因此我们建议在后台运行此脚本:
scripts/download_all_data.sh <DOWNLOAD_DIR> > download.log 2> download_all.log &-
**注意:下载目录
<DOWNLOAD_DIR>不应是AlphaFold仓库目录中的子目录。**如果是,Docker构建将会很慢,因为大型数据库将被复制到docker构建上下文中。 -
�可以使用精简数据库运行AlphaFold;请参阅完整文档。
-
-
通过运行以下命令检查AlphaFold是否能够使用GPU:
docker run --rm --gpus all nvidia/cuda:11.0-base nvidia-smi此命令的输出应显示您的GPU列表。如果没有,请检查在设置NVIDIA Container Toolkit时是否正确执行了所有步骤,或查看以下NVIDIA Docker问题。
如果您希望使用Singularity(HPC系统上常见的容器化平台)运行AlphaFold,我们建议使用一些第三方Singularity设置,如https://github.com/deepmind/alphafold/issues/10 或 https://github.com/deepmind/alphafold/issues/24 中所链接的。
-
构建Docker镜像:
docker build -f docker/Dockerfile -t alphafold .如果遇到以下错误:
W: GPG error: https://developer.download.nvidia.com/compute/cuda/repos/ubuntu1804/x86_64 InRelease: The following signatures couldn't be verified because the public key is not available: NO_PUBKEY A4B469963BF863CC E: The repository 'https://developer.download.nvidia.com/compute/cuda/repos/ubuntu1804/x86_64 InRelease' is not signed.请使用 https://github.com/deepmind/alphafold/issues/463#issuecomment-1124881779 中描述的解决方法。
-
安装
run_docker.py依赖项。注意:您可以选择创建Python虚拟环境以防止与系统的Python环境发生冲突。pip3 install -r docker/requirements.txt -
确保输出目录存在(默认为
/tmp/alphafold)并且您有足够的权限写入其中。 -
运行
run_docker.py,指向包含您希望预测结构的蛋白质序列的FASTA文件(--fasta_paths参数)。AlphaFold将在--max_template_date参数指定的日期之前搜索可用的模板;这可用于在建模过程中避免某些模板。--data_dir是下载的遗传数据库目录,--output_dir是输出目录的绝对路径。python3 docker/run_docker.py \ --fasta_paths=your_protein.fasta \ --max_template_date=2022-01-01 \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir -
运行完成后,输出目录将包含目标蛋白质的预测结构。请查看下面的文档以获取其他选项和故障排除提示。
遗传数据库
此步骤需要在您的机器上安装aria2c。
AlphaFold需要多个遗传(序列)数据库才能运行:
- BFD,
- MGnify,
- PDB70,
- PDB(mmCIF格式的结构),
- PDB seqres – 仅用于AlphaFold-Multimer,
- UniRef30(前身为UniClust30),
- UniProt – 仅用于AlphaFold-Multimer,
- UniRef90。
我们提供了一个脚本scripts/download_all_data.sh,可用于下载和设置所有这些数据库:
-
推荐的默认设置:
scripts/download_all_data.sh <DOWNLOAD_DIR>将下载完整的数据库。
-
使用
reduced_dbs参数:scripts/download_all_data.sh <DOWNLOAD_DIR> reduced_dbs将下载数据库的精简版本,用于
reduced_dbs数据库预设。在之后的AlphaFold运行中,这应与相应的AlphaFold参数--db_preset=reduced_dbs一起使用(请参阅AlphaFold参数部分)。 :ledger: 注意:下载目录<DOWNLOAD_DIR>不应该是 AlphaFold 仓库目录的子目录。 如果是的话,Docker 构建过程会很慢,因为大型数据库会在镜像创建过程中被复制。
我们没有提供 CASP14 中使用的完全相同的数据库版本 – 请参阅关于可重复性的说明。为了提高速度,一些数据库是镜像的,请参阅镜像数据库。
:ledger: 注意:完整数据库的总下载大小约为 556 GB,解压后的总大小为 2.62 TB。请确保您有足够的硬盘空间、带宽和时间来下载。我们建议使用 SSD 以获得更好的基因搜索性能。
:ledger: 注意:如果下载目录和数据集没有完全的读写权限,可能会导致 MSA 工具出错,并显示不透明(外部)的错误消息。请确保应用了所需的权限,例如使用 sudo chmod 755 --recursive "$DOWNLOAD_DIR" 命令。
download_all_data.sh 脚本还将下载模型参数文件。脚本完成后,您应该有以下目录结构:
$DOWNLOAD_DIR/ # 总计: ~ 2.62 TB (下载: 556 GB)
bfd/ # ~ 1.8 TB (下载: 271.6 GB)
# 6 个文件
mgnify/ # ~ 120 GB (下载: 67 GB)
mgy_clusters_2022_05.fa
params/ # ~ 5.3 GB (下载: 5.3 GB)
# 5 个 CASP14 模型,
# 5 个 pTM 模型,
# 5 个 AlphaFold-Multimer 模型,
# LICENSE,
# = 16 个文件
pdb70/ # ~ 56 GB (下载: 19.5 GB)
# 9 个文件
pdb_mmcif/ # ~ 238 GB (下载: 43 GB)
mmcif_files/
# 约 199,000 个 .cif 文件
obsolete.dat
pdb_seqres/ # ~ 0.2 GB (下载: 0.2 GB)
pdb_seqres.txt
small_bfd/ # ~ 17 GB (下载: 9.6 GB)
bfd-first_non_consensus_sequences.fasta
uniref30/ # ~ 206 GB (下载: 52.5 GB)
# 7 个文件
uniprot/ # ~ 105 GB (下载: 53 GB)
uniprot.fasta
uniref90/ # ~ 67 GB (下载: 34 GB)
uniref90.fasta
只有在下载完整数据库时才会下载 bfd/,只有在下载精简数据库时才会下载 small_bfd/。
模型参数
虽然 AlphaFold 代码以 Apache 2.0 许可证发布,但 AlphaFold 参数和 CASP15 预测数据是根据 CC BY 4.0 许可条款提供的。更多详情请参阅下方的免责声明。
AlphaFold 参数可从 https://storage.googleapis.com/alphafold/alphafold_params_2022-12-06.tar 获取,并作为 scripts/download_all_data.sh 脚本的一部分下载。这个脚本将下载以下参数:
- 5 个在 CASP14 期间使用的模型,这些模型已经过广泛验证,确保结构预测质量(详见 Jumper 等人 2021 年文章,补充方法 1.12)。
- 5 个 pTM 模型,这些模型经过微调,可以在结构预测的同时产生 pTM(预测的 TM-score)和 PAE(预测的对齐误差)值(详见 Jumper 等人 2021 年文章,补充方法 1.9.7)。
- 5 个 AlphaFold-Multimer 模型,可以在结构预测的同时产生 pTM 和 PAE 值。
更新现有安装
如果您有之前的版本,可以选择从头开始完全重新安装(删除所有内容并重新运行设置),或者进行增量更新,这会显著加快速度,但需要更多工作。请确保按照以下步骤的确切顺序执行:
- 更新代码。
- 进入克隆的 AlphaFold 仓库目录,运行
git fetch origin main以获取所有代码更新。
- 进入克隆的 AlphaFold 仓库目录,运行
- 更新 UniProt、UniRef、MGnify 和 PDB seqres 数据库。
- 删除
<DOWNLOAD_DIR>/uniprot。 - 运行
scripts/download_uniprot.sh <DOWNLOAD_DIR>。 - 删除
<DOWNLOAD_DIR>/uniclust30。 - 运行
scripts/download_uniref30.sh <DOWNLOAD_DIR>。 - 删除
<DOWNLOAD_DIR>/uniref90。 - 运行
scripts/download_uniref90.sh <DOWNLOAD_DIR>。 - 删除
<DOWNLOAD_DIR>/mgnify。 - 运行
scripts/download_mgnify.sh <DOWNLOAD_DIR>。 - 删除
<DOWNLOAD_DIR>/pdb_mmcif。为了确保 PDB SeqRes 和 PDB 的日期完全相同,必须执行此步骤。如果不执行此步骤,在运行 AlphaFold-Multimer 时搜索模板可能会出错。 - 运行
scripts/download_pdb_mmcif.sh <DOWNLOAD_DIR>。 - 运行
scripts/download_pdb_seqres.sh <DOWNLOAD_DIR>。
- 删除
- 更新模型参数。
- 删除
<DOWNLOAD_DIR>/params中的旧模型参数。 - 使用
scripts/download_alphafold_params.sh <DOWNLOAD_DIR>下载新的模型参数。
- 删除
- 按照运行 AlphaFold 的说明进行操作。
使用已弃用的模型权重
要使用已弃用的 v2.2.0 AlphaFold-Multimer 模型权重:
- 将
scripts/download_alphafold_params.sh中的SOURCE_URL更改为https://storage.googleapis.com/alphafold/alphafold_params_2022-03-02.tar,并下载旧参数。 - 将
config.py中 multimerMODEL_PRESETS的_v3改为_v2。
要使用已弃用的 v2.1.0 AlphaFold-Multimer 模型权重:
- 将
scripts/download_alphafold_params.sh中的SOURCE_URL更改为https://storage.googleapis.com/alphafold/alphafold_params_2022-01-19.tar,并下载旧参数。 - 删除
config.py中 multimerMODEL_PRESETS的_v3。
运行 AlphaFold
运行 AlphaFold 最简单的方法是使用提供的 Docker 脚本。 这在 Google Cloud 上进行了测试,使用的机器配置为 nvidia-gpu-cloud-image,12 个 vCPU,85 GB RAM,100 GB 引导磁盘,数据库存储在额外的 3 TB 磁盘上,以及一个 A100 GPU。对于您的第一次运行,请按照安装和运行您的第一个预测部分的说明进行操作。
-
默认情况下,Alphafold 将尝试使用所有可见的 GPU 设备。要使用子集,请使用
--gpu_devices标志指定逗号分隔的 GPU UUID 或索引列表。有关详细信息,请参阅GPU 枚举。 -
您可以通过添加
--model_preset=标志来控制要运行的 AlphaFold 模型。我们提供以下模型:-
monomer:这是在 CASP14 中使用的原始模型,没有集成。
-
monomer_casp14:这是在 CASP14 中使用的原始模型,设置
num_ensemble=8,与我们的 CASP14 配置相匹配。这主要是为了可重现性而提供的,因为它在计算上要贵 8 倍,而精度提升有限(CASP14 域上平均 GDT 提升 +0.1)。 -
monomer_ptm:这是用 pTM 头进行微调的原始 CASP14 模型,提供了成对的置信度度量。它的准确性略低于普通单体模型。
-
multimer:这是 AlphaFold-Multimer 模型。要使用此模型,请提供多序列 FASTA 文件。此外,应该已经下载了 UniProt 数据库。
-
-
您可以通过在运行命令中添加
--db_preset=reduced_dbs或--db_preset=full_dbs来控制 MSA 速度/质量的权衡。我们提供以下预设:- reduced_dbs:此预设针对速度和较低的硬件要求进行�了优化。它使用 BFD 数据库的精简版本运行。它需要 8 个 CPU 核心(vCPU),8 GB RAM 和 600 GB 磁盘空间。
- full_dbs:这将使用CASP14中使用的所有遗传数据库运行。
使用 monomer 模型预设和 reduced_dbs 数据预设运行上述命令将如下所示:
python3 docker/run_docker.py \ --fasta_paths=T1050.fasta \ --max_template_date=2020-05-14 \ --model_preset=monomer \ --db_preset=reduced_dbs \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir
-
在生成预测模型后,AlphaFold会运行一个松弛步骤来改善局部几何结构。默认情况下,只对最佳模型(按pLDDT评分)进行松弛(
--models_to_relax=best),但也可以对所有模型(--models_to_relax=all)或不对任何模型(--models_to_relax=none)进行松弛。 -
松弛步骤可以在GPU上运行(更快,但可能不太稳定)或CPU上运行(慢,但稳定)。这可以通过
--enable_gpu_relax=true(默认)或--enable_gpu_relax=false来控制。 -
AlphaFold可以通过
--use_precomputed_msas=true选项重复使用相同序列的MSA(多序列比对);这对于尝试不同的AlphaFold参数很有用。此选项假设输出目录中存在第一次AlphaFold运行生成的目录结构,并且蛋白质序列相同。
运行AlphaFold-Multimer
所有步骤与运行单体系统相同,但您需要
- 提供包含多个序列的输入fasta文件,
- 设置
--model_preset=multimer,
以下是折叠蛋白质复合物 multimer.fasta 的示例:
python3 docker/run_docker.py \ --fasta_paths=multimer.fasta \ --max_template_date=2020-05-14 \ --model_preset=multimer \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir
默认情况下,多聚体系统将为每个模型运行5个种子(共25个预测)。如果您希望稍微降��低准确性以减少运行时间,可以为每个模型运行单个种子。这可以通过 --num_multimer_predictions_per_model 标志来实现,例如,设置为 --num_multimer_predictions_per_model=1 以为每个模型运行单个种子。
AlphaFold预测速度
下表报告了各种长度蛋白质的预测运行时间。我们仅测量了三次循环的未松弛结构预测,同时排除了MSA和模板搜索的运行时间。当使用 --benchmark=true 运行 docker/run_docker.py 时,这个运行时间会存储在 timings.json 中。所有运行时间都来自单个NVIDIA A100 GPU。通过增加 alphafold/model/config.py 中的 global_config.subbatch_size,可以提高A100上较小结构的预测速度。
| 残基数 | 预测时间(秒) |
|---|---|
| 100 | 4.9 |
| 200 | 7.7 |
| 300 | 13 |
| 400 | 18 |
| 500 | 29 |
| 600 | 36 |
| 700 | 53 |
| 800 | 60 |
| 900 | 91 |
| 1,000 | 96 |
| 1,100 | 140 |
| 1,500 | 280 |
| 2,000 | 450 |
| 2,500 | 969 |
| 3,000 | 1,240 |
| 3,500 | 2,465 |
| 4,000 | 5,660 |
| 4,500 | 12,475 |
| 5,000 | 18,824 |
示例
以下是在不同情况下使用AlphaFold的示例。
折叠单体
假设我们有一个序列为 <SEQUENCE> 的单体。输入的fasta文件应为:
>sequence_name <SEQUENCE>
然后运行以下命令:
python3 docker/run_docker.py \ --fasta_paths=monomer.fasta \ --max_template_date=2021-11-01 \ --model_preset=monomer \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir
折叠同源多聚体
假设我们有一个包含3个相同序列 <SEQUENCE> 的同源多聚体。输入的fasta文件应为:
>sequence_1 <SEQUENCE> >sequence_2 <SEQUENCE> >sequence_3 <SEQUENCE>
然后运行以下命令:
python3 docker/run_docker.py \ --fasta_paths=homomer.fasta \ --max_template_date=2021-11-01 \ --model_preset=multimer \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir
折叠异源多聚体
假设我们有一个A2B3异源多聚体,即2个 <SEQUENCE A> 和3个 <SEQUENCE B>。输入的fasta文件应为:
>sequence_1 <SEQUENCE A> >sequence_2 <SEQUENCE A> >sequence_3 <SEQUENCE B> >sequence_4 <SEQUENCE B> >sequence_5 <SEQUENCE B>
然后运行以下命令:
python3 docker/run_docker.py \ --fasta_paths=heteromer.fasta \ --max_template_date=2021-11-01 \ --model_preset=multimer \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir
连续折叠多个单体
假设我们有两个单体,monomer1.fasta 和 monomer2.fasta。
我们可以使用以下命令连续折叠两者:
python3 docker/run_docker.py \ --fasta_paths=monomer1.fasta,monomer2.fasta \ --max_template_date=2021-11-01 \ --model_preset=monomer \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir
连续折叠多个多聚体
假设我们有两个多聚体,multimer1.fasta 和 multimer2.fasta。
我们可以使用以下命令连续折叠两者:
python3 docker/run_docker.py \ --fasta_paths=multimer1.fasta,multimer2.fasta \ --max_template_date=2021-11-01 \ --model_preset=multimer \ --data_dir=$DOWNLOAD_DIR \ --output_dir=/home/user/absolute_path_to_the_output_dir
AlphaFold输出
输出将保存在通过 run_docker.py 的 --output_dir 标志提供的目录的子目录中(默认为 /tmp/alphafold/)。输出包括计算得到的MSA、未松弛结构、松弛结构、排序结构、原始模型输出、预测元数据和部分时间。--output_dir 目录将具有以下结构:
<target_name>/
features.pkl
ranked_{0,1,2,3,4}.pdb
ranking_debug.json
relax_metrics.json
relaxed_model_{1,2,3,4,5}.pdb
result_model_{1,2,3,4,5}.pkl
timings.json
unrelaxed_model_{1,2,3,4,5}.pdb
msas/
bfd_uniref_hits.a3m
mgnify_hits.sto
uniref90_hits.sto
每个输出文件的内容如下:
-
features.pkl- 一个包含模型用于生成结构的输入特征NumPy数组的pickle文件。 -
unrelaxed_model_*.pdb- 一个包含模型直接输出的预测结构的PDB格式文本文件。 -
relaxed_model_*.pdb- 一个包含对未放松的结构预测进行Amber放松程序后的预测结构的PDB格式文本文件(详见Jumper等人2021年文章,补充方法1.8.6)。 -
ranked_*.pdb- 一个包含按模型置信度重新排序后的预测结构的PDB格式文本文件。这里ranked_i.pdb应包含第(i+1)高置信度的预测(因此ranked_0.pdb具有最高置信度)。为了对模型置信度排序,我们使用预测的LDDT(pLDDT)分数(详见Jumper等人2021年文章,补充方法1.9.6)。如果--models_to_relax=all,则所有排序的结构都会被放松。如果--models_to_relax=best,则只有ranked_0.pdb会被放松(其余未放松)。如果--models_to_relax=none,则排序的结构都未放松。 -
ranking_debug.json- 一个JSON格式的文本文件,包含用于模型排序的pLDDT值,以及回溯到原始模型名称的映射。 -
relax_metrics.json- 一个JSON格式的文本文件,包含放松指标,例如剩余违例。 -
timings.json- 一个JSON格式的文本文件,包含运行AlphaFold流程各部分所用的时间。 -
msas/- 一个包含用于构建输入MSA的各种遗传工具匹配文件的目录。 -
result_model_*.pkl- 一个包含模型直接生成的各种NumPy数组嵌套字典的pickle文件。除了结构模块的输出外,还包括辅助输出,如:- 距离图(
distogram/logits包含形状为[N_res, N_res, N_bins]的NumPy数组,distogram/bin_edges包含bin的定义)。 - 每个残基的pLDDT分数(
plddt包含形状为[N_res]的NumPy数组,可能的值范围从0到100,其中100表示最有信心)。这可用于识别预测置信度高的序列区域,或作为跨残基平均的每个目标的整体置信度分数。 - 仅在使用pTM模型时存在:预测的TM-score(
ptm字段包含一个标量)。作为全局叠加度量的预测器,该分数还旨在评估模型对整体结构域打包的置信度。 - 仅在使用pTM模型时存在:预测的成对对齐误差(
predicted_aligned_error包含形状为[N_res, N_res]的NumPy数组,可能的值范围从0到max_predicted_aligned_error,其中0表示最有信心)。这可用于可视化结构内的结构域打包置信度。
- 距离图(
pLDDT置信度指标存储在输出PDB文件的B因子字段中(尽管与B因子不同,较高的pLDDT更好,因此在用于分子替换等任务时必须小心)。
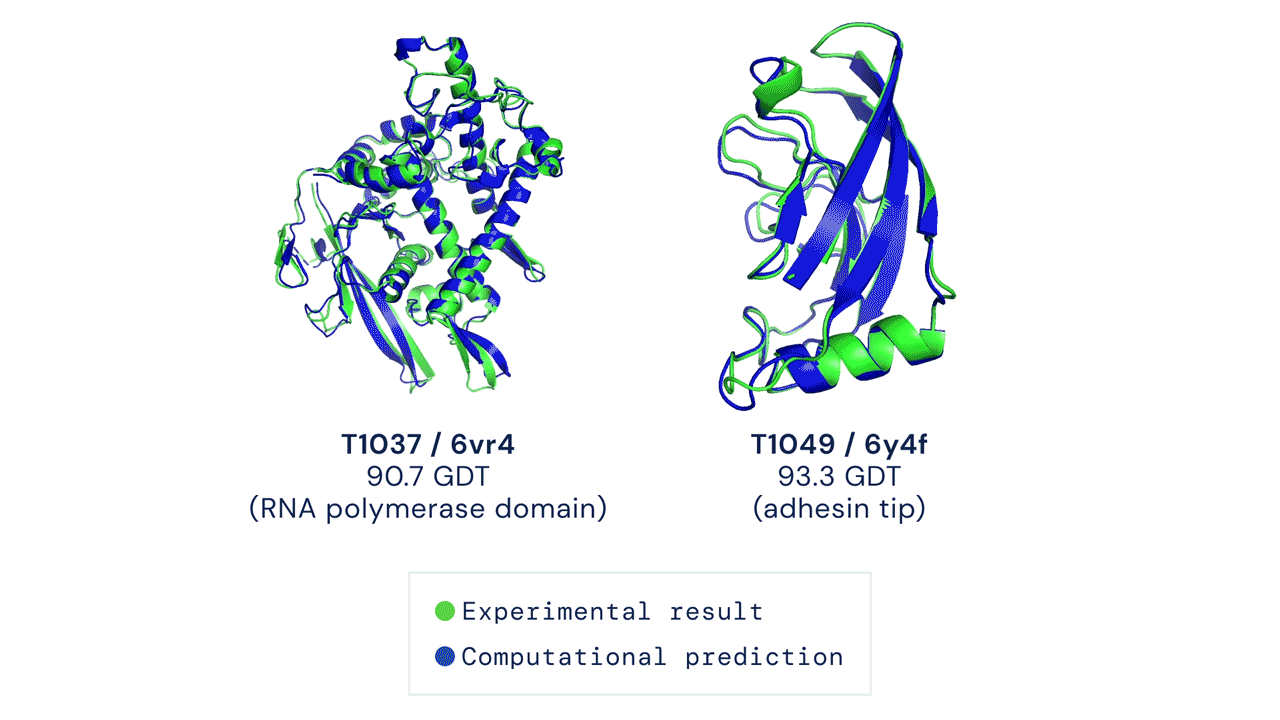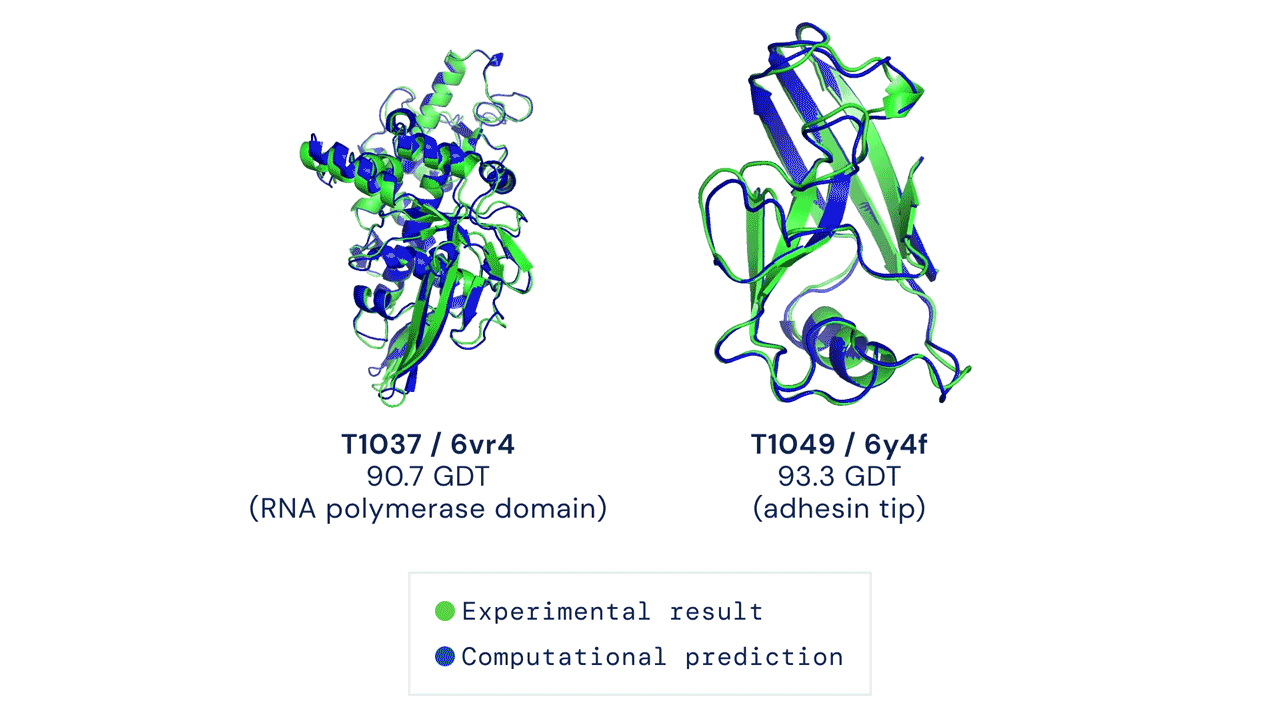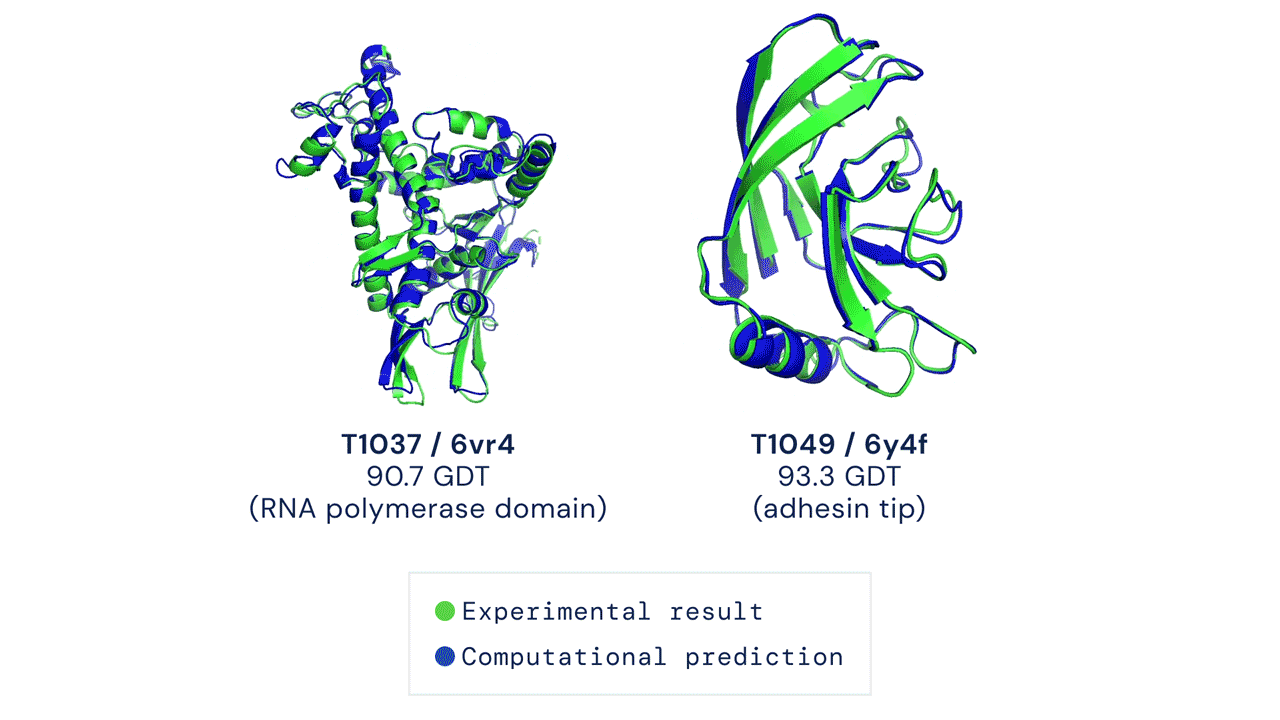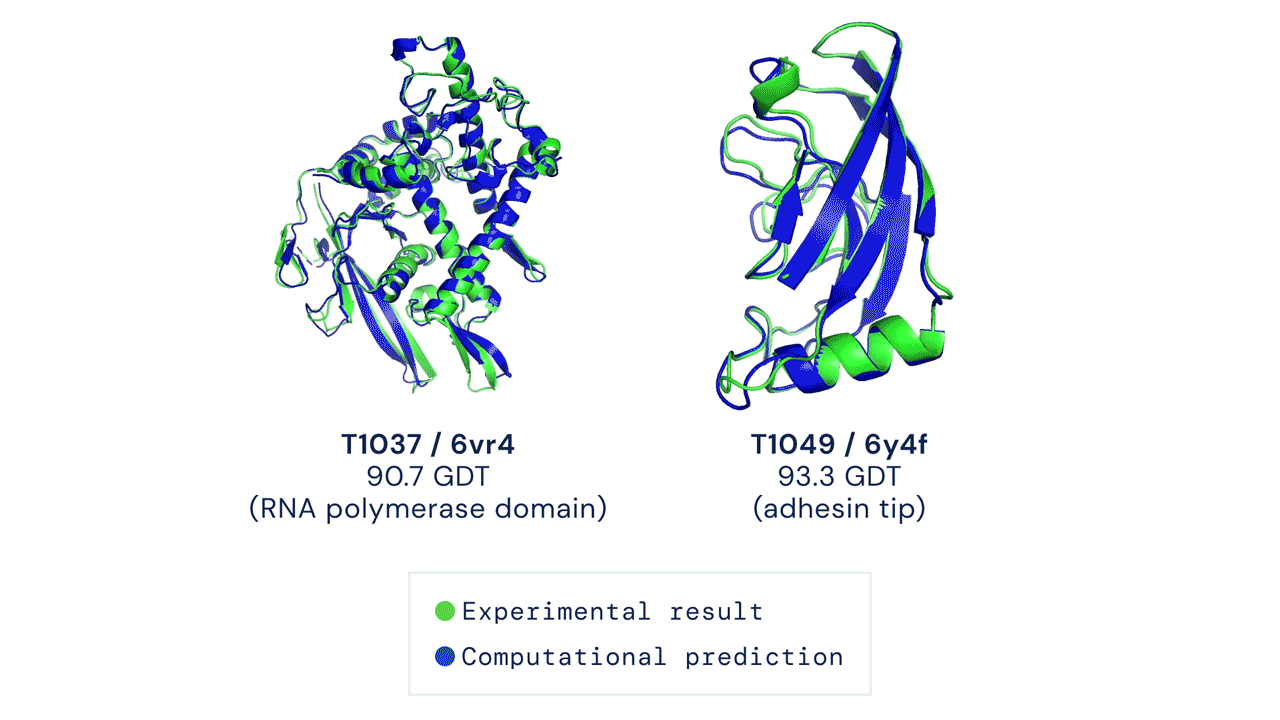
该代码经测试,在CASP14测试集上的5个模型预测中,使用pLDDT排序的平均top-1准确率与我们的结果相匹配(一些CASP目标使用了早期版本的AlphaFold运行,一些进行了人工干预;详见我们即将发表的论文)。一些目标如T1064可能在随机种子上也有较高的单次运行方差。
推断多个蛋白质
提供的推断脚本针对预测单个蛋白质的结构进行了优化,它会编译神经网络以专门适应序列、MSA和模板的确切大小。对于大型蛋白质,编译时间是运行时间的一小部分,但对于小型蛋白质或如果多序列比对已经预先计算好,它可能变得更加重要。在批量推断的情况下,使用我们的make_fixed_size函数将输入填充到统一大小可能更有意义,从而减少所需的编译次数。
我们不提供批量推断脚本,但应该可以在RunModel.predict方法的基础上直接开发,并配合预计算多序列比对的并行系统。或者,可以重复运行此脚本,只会产生适度的开销。
关于CASP14可重复性的说明
AlphaFold对少数蛋白质的输出具有高度的运行间变异性,并可能受到输入数据变化的影响。CASP14目标T1064是一个显著的例子;最近提交的大量SARS-CoV-2相关序列显著改变了其MSA。这种可变性在一定程度上通过模型选择过程得到缓解;运行5个模型并选择最有信心的一个。
为了尽可能准确地重现我们CASP14系统的结果,你必须使用我们在CASP中使用的相同数据库版本。这些可能与我们脚本下载的默认版本不匹配。
对于遗传学:
对于模板:
- PDB: (下载于2020-05-14)
- PDB70: 2020-05-13
模板的另一种选择是使用最新的PDB和PDB70,但传递标志--max_template_date=2020-05-14,这将模板限制为CASP14开始时可用的结构。
引用本工作
如果您使用本包中的代码或数据,请引用:
@Article{AlphaFold2021, author = {Jumper, John and Evans, Richard and Pritzel, Alexander and Green, Tim and Figurnov, Michael and Ronneberger, Olaf and Tunyasuvunakool, Kathryn and Bates, Russ and {\v{Z}}{\'\i}dek, Augustin and Potapenko, Anna and Bridgland, Alex and Meyer, Clemens and Kohl, Simon A A and Ballard, Andrew J and Cowie, Andrew and Romera-Paredes, Bernardino and Nikolov, Stanislav and Jain, Rishub and Adler, Jonas and Back, Trevor and Petersen, Stig and Reiman, David and Clancy, Ellen and Zielinski, Michal and Steinegger, Martin and Pacholska, Michalina and Berghammer, Tamas and Bodenstein, Sebastian and Silver, David and Vinyals, Oriol and Senior, Andrew W and Kavukcuoglu, Koray and Kohli, Pushmeet and Hassabis, Demis}, journal = {Nature}, title = {Highly accurate protein structure prediction with {AlphaFold}}, year = {2021}, volume = {596}, number = {7873}, pages = {583--589}, doi = {10.1038/s41586-021-03819-2} }
此外,如果您使用AlphaFold-Multimer模式,请引用:
@article {AlphaFold-Multimer2021, author = {Evans, Richard 和 O'Neill, Michael 和 Pritzel, Alexander 和 Antropova, Natasha 和 Senior, Andrew 和 Green, Tim 和 Žídek, Augustin 和 Bates, Russ 和 Blackwell, Sam 和 Yim, Jason 和 Ronneberger, Olaf 和 Bodenstein, Sebastian 和 Zielinski, Michal 和 Bridgland, Alex 和 Potapenko, Anna 和 Cowie, Andrew 和 Tunyasuvunakool, Kathryn 和 Jain, Rishub 和 Clancy, Ellen 和 Kohli, Pushmeet 和 Jumper, John 和 Hassabis, Demis}, journal = {bioRxiv}, title = {使用AlphaFold-Multimer预测蛋白质复合物}, year = {2021}, elocation-id = {2021.10.04.463034}, doi = {10.1101/2021.10.04.463034}, URL = {https://www.biorxiv.org/content/early/2021/10/04/2021.10.04.463034}, eprint = {https://www.biorxiv.org/content/early/2021/10/04/2021.10.04.463034.full.pdf}, }
社区贡献
由社区提供的Colab笔记本(请注意,这些笔记本可能与我们完整的AlphaFold系统有所不同,我们没有验证它们的准确性):
- Martin Steinegger、Sergey Ovchinnikov和Milot Mirdita提供的ColabFold AlphaFold2笔记本,该笔记本使用Södinglab托管的API,基于MMseqs2服务器(Mirdita等人,2019年,Bioinformatics)进行多序列比对。
致谢
AlphaFold与以下独立库和软件包进行通信和/或引用:
- Abseil
- Biopython
- Chex
- Colab
- Docker
- HH Suite
- HMMER Suite
- Haiku
- Immutabledict
- JAX
- Kalign
- matplotlib
- ML Collections
- NumPy
- OpenMM
- OpenStructure
- pandas
- pymol3d
- SciPy
- Sonnet
- TensorFlow
- Tree
- tqdm
我们感谢所有这些项目的贡献者和维护者!
联系我们
如果您有任何本概述中未涉及的问题,请通过alphafold@deepmind.com联系AlphaFold团队。
我们很乐意听到您的反馈,并了解AlphaFold如何在您的研究中发挥作用。请通过alphafold@deepmind.com与我们分享您的故事。
许可和免责声明
这不是Google官方支持的产品。
版权所有 2022 DeepMind Technologies Limited。
AlphaFold代码许可
根据Apache许可证2.0版("许可证")获得许可;除非符合许可证,否则您不得使用此文件。您可以在https://www.apache.org/licenses/LICENSE-2.0获取许可证的副本。
除非适用法律要求或书面同意,根据许可证分发的软件是基于"按原样"的基础分发的,没有任何明示或暗示的担保或条件。请参阅许可证以了解许可证下的特定语言和限制。
模型参数许可
AlphaFold参数根据知识共享署名4.0国际(CC BY 4.0)许可提供。您可以在以下网址找到详细信息:https://creativecommons.org/licenses/by/4.0/legalcode
第三方软件
使用上述致谢部分中提到的第三方软件、库或代码可能受单独的条款和条件�或许可条款的约束。您对第三方软件、库或代码的使用受任何此类条款的约束,您应该在使用之前检查您是否可以遵守任何适用的限制或条款和条件。
镜像数据库
以下数据库已由DeepMind镜像,并可参考以下内容:
-
BFD(未修改),由Steinegger M.和Söding J.提供,根据知识共享署名-相同方式共享4.0国际许可提供。
-
BFD(修改版),由Steinegger M.和Söding J.提供,由DeepMind修改,根据知识共享署名-相同方式共享4.0国际许可提供。详情请参见AlphaFold蛋白质组论文的方法部分。
-
Uniref30: v2021_03(未修改),由Mirdita M.等人提供,根据知识共享署名-相同方式共享4.0国际许可提供。
-
MGnify: v2022_05(未修改),由Mitchell AL等人提供,免除所有版权限制,根据CC0 1.0通用(CC0 1.0)公共领域贡献完全免费提供,可用于非商业和商业用途。
编辑推荐精选


扣子-AI办公
职场AI,就用扣子
AI办公助手,复杂任务高效处理。办公效率低?扣子空间AI助手支持播客生成、PPT制作、网页开发及报告写作,覆盖科研、商业、舆情等领域的专家Agent 7x24小时响应,生活工作无缝切换,提升50%效率!


堆友
多风格AI绘画神器
堆友平台由阿里巴巴设计团队创建,作为一款AI驱动的设计工具,专为设计师提供一站式增长服务。功能覆盖海量3D素材、AI绘画、实时渲染以及专业抠图,显著提升设计品质和效率。平台不仅提供工具,还是一个促进创意交流和个人发展的空间,界面友好,适合所有级别的设计师和创意工作者。


码上飞
零代码AI应用开发平台
零代码AI应用开发平台,用户只需一句话简单描述需求,AI能自动生成小程序、APP或H5网页应用,无需编写代码。

Vora
免费创建高清无水印Sora视频
Vora是一个免费创建高清无水印Sora视频的AI工具


Refly.AI
最适合小白的AI自动化工作流平台
无需编码,轻松生成可复用、可变现的AI自动化工作流


酷表ChatExcel
大模型驱动的Excel数据处理工具
基于大模型交互的表格处理系统,允许用户通过对话方式完成数据整理和可视化分析。系统采用机器学习算法解析用户指令,自动执行排序、公式计算和数据透视等操作,支持多种文件格式导入导出。数据处理响应速度保持在0.8秒以内,支持超过100万行数据的即时分析。


TRAE编程
AI辅助编程,代码自动修复
Trae是一种自适应的集成开发环境(IDE),通过自动化和多元协作改变开发流程。利用Trae,团队能够更快速、精确地编写和部署代码,从而提高编程效率和项目交付速度。Trae具备上下文感知和代码自动完成功能,是提升开发效率的理想工具。


AIWritePaper论文写作
AI论文写作指导平台
AIWritePaper论文写作是一站式AI论文写作辅助工具,简化了选题、文献检索至论文撰写的整个过程。通过简单设定,平台可快速生成高质量论文大纲和全文,配合图表、参考文献等一应俱全,同时提供开题报告和答辩PPT等增值服务,保障数据安全,有效提升写作效率和论文质量。

博思AIPPT
AI一键生成PPT,就用博思AIPPT!
博思AIPPT,新一代的AI生成PPT平台,支持智能生成PPT、AI美化PPT、文本&链接生成PPT、导入Word/PDF/Markdown文档生成PPT等,内置海量精美PPT模板,涵盖商务、教育、科技等不同风格,同时针对每个页面提供多种版式,一键自适应切换,完美适配各种办公场景。


潮际好麦
AI赋能电商视觉革命,一站式智能商拍平台
潮际好麦深耕服装行业,是国内AI试衣效果最好的软件。使用先进AIGC能力为电商卖家批量提供优质的、低成本的商拍图。合作品牌有Shein、Lazada、安踏、百丽等65个国内外头部�品牌,以及国内10万+淘宝、天猫、京东等主流平台的品牌商家,为卖家节省将近85%的出图成本,提升约3倍出图效率,让品牌能够快速上架。
推荐工具精选




AI云服务特惠
懂AI专属折扣关注微信公众号
最新AI工具、AI资讯
独家AI资源、AI项目落地

微信扫一扫关注公众号