AlphaFold 3的PyTorch实现:蛋白质结构预测的突破性进展
 Ray
RayAlphaFold 3:蛋白质结构预测的革命性进展
AlphaFold 3是人工智能在生物学领域的一项重大突破,它为准确预测生物分子的结构和相互作用开辟了新的可能。本文将深入探讨AlphaFold 3的PyTorch实现,解析其核心技术和创新点,为研究者和开发者提供宝贵的参考。
AlphaFold 3的核心创新
AlphaFold 3相比于前代模型有以下几个关键的创新:
-
更广泛的应用范围:不仅可以预测蛋白质结构,还能预测核酸、配体等多种生物分子的结构和相互作用。
-
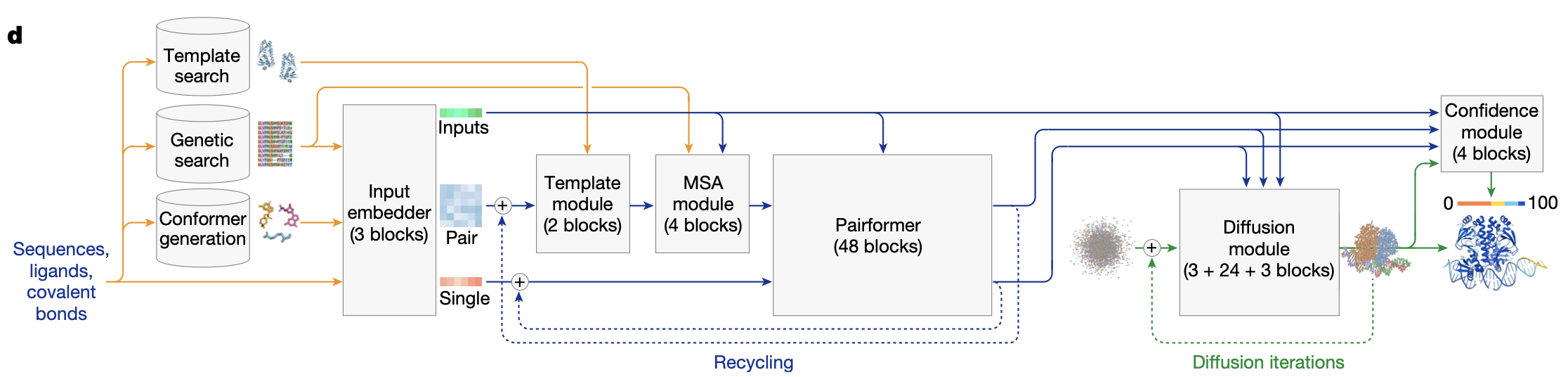
改进的网络架构:引入了PairFormer模块替代了EvoFormer,能更有效地处理配对表示和单独表示。
-
扩散模型的应用:直接在原子坐标上操作,通过去噪任务学习蛋白质结构的多尺度特征。
-
交叉蒸馏技术:通过enriching训练数据来抑制模型的幻觉问题。
-
置信度预测:能够预测原子层面和配对层面的误差,提高结果的可靠性。
这些创新使AlphaFold 3在准确性和适用范围上都有了显著提升。
PyTorch实现的关键模块
在PyTorch的实现中,主要包含以下几个核心模块:
-
输入嵌入模块:处理序列、MSA和模板信息。
-
PairFormer模块:替代了EvoFormer,处理配对表示和单独表示。
-
扩散模块:直接在原子坐标上进行操作。
-
置信度预测模块:预测结构的误差。
下面是一个简化的使用示例:
import torch from alphafold3_pytorch import Alphafold3 # 初始化模型 alphafold3 = Alphafold3( dim_atom_inputs = 77, dim_template_feats = 108 ) # 准备输入数据 seq_len = 16 atom_inputs = torch.randn(2, seq_len, 77) atompair_inputs = torch.randn(2, seq_len, seq_len, 5) # ... 其他输入 ... # 训练 loss = alphafold3( num_recycling_steps = 2, atom_inputs = atom_inputs, atompair_inputs = atompair_inputs, # ... 其他参数 ... ) loss.backward() # 推理 sampled_atom_pos = alphafold3( num_recycling_steps = 4, num_sample_steps = 16, atom_inputs = atom_inputs, atompair_inputs = atompair_inputs, # ... 其他参数 ... )
数据准备和处理
AlphaFold 3的训练需要大量高质量的数据。主要的数据来源是蛋白质数据库(PDB)。数据处理流程包括:
- 下载PDB数据
- 数据过滤
- 数据聚类
这个过程确保了训练数据的质量和多样性。详细的数据处理脚本和步骤可以在项目的GitHub仓库中找到。
模型训练和推理
AlphaFold 3的训练过程涉及多个损失函数,包括距离图预测、原子位置预测等。训练时需要大量的计算资源,通常需要使用GPU集群。
在推理阶段,模型会生成多个样本,然后根据置信度选择最佳结果。这种方法能够提高预测的稳定性和准确性。
社区贡献和未来展望
AlphaFold 3的开源实现得到了广泛的社区支持。许多研究者和开发者为项目贡献了宝贵的改进和功能扩展。这种开放合作的模式极大地推动了项目的发展。
未来,AlphaFold 3还有很大的发展空间:
- 进一步提高对大型复合物的预测能力
- 改进对动态结构的预测
- 扩展到更多类型的生物分子
- 提高计算效率,使其能在更普及的硬件上运行
结论
AlphaFold 3代表了蛋白质结构预测领域的一个重要里程碑。它的PyTorch实现为研究者和开发者提供了一个强大的工具,有望加速生物学和药物研发等领域的创新。随着社区的不断贡献和技术的持续进步,我们可以期待看到更多令人兴奋的应用和突破。
参考资源
通过深入了解AlphaFold 3及其PyTorch实现,研究者和开发者可以更好地利用这一强大工具,推动生命科学研究的边界。让我们共同期待AlphaFold 3带来的更多突破性进展!
编辑推荐精选


音述AI
全球首个AI音乐社区
音述AI是全球首个AI音乐社区,致力让每个人都能用音乐表达自我。音述AI提供零门槛AI创作工具,独创GETI法则帮助用户精准定义音乐风格,AI润色功能支持自动优化作品质感。音述AI支持交流讨论、二次创作与价值变现。针对中文用户的语言习惯与文化背景进行专门优化,支持国风融合、C-pop等本土音乐标签,让技术更好地承载人文表达。


QoderWork
阿里Qoder团队推出的桌面端AI智能体
QoderWork 是阿里推出的本地优先桌面 AI 智能体,适配 macOS14+/Windows10+,以自然语言交互实现文件管理、数据分析、AI 视觉生成、浏览器自动化等办公任务,自主拆解执行复杂工作流,数据本地运行零上传,技能市场可无限扩展,是高效的 Agentic 生产力办公助手。


lynote.ai
一站式搞定所有学习需求
不再被海量信息淹没,开始真正理解知识。Lynote 可摘要 YouTube 视频、PDF、文章等内容。即时创建笔记,检测 AI 内容并下载资料,将您的学习效率提升 10 倍。


AniShort
为AI短剧协作而生
专为AI短剧协作而生的AniShort正式发布,深度重构AI短剧全流程生产模式,整合创意策划、制作执行、实时协作、在线审片、资产复用等全链路功能,独创无限画布、双轨并行工业化工作流与Ani智能体助手,集成多款主流AI大模型,破解素材零散、版本混乱、沟通低效等行业痛点,助力3人团队效率提升800%,打造标准化、可追溯的AI短剧量产体系,是AI短剧团队协同创作、提升制作效率的核心工具。


seedancetwo2.0
能听懂你表达的视频模型
Seedance two是基于seedance2.0的中国大模型,支持图�像、视频、音频、文本四种模态输入,表达方式更丰富,生成也更可控。


nano-banana纳米香蕉中文站
国内直接访问,限时3折
输入简单文字,生成想要的图片,纳米香蕉中文站基于 Google 模型的 AI 图片生成网站,支持文字生图、图生图。官网价格限时3折活动


扣子-AI办公
职场AI,就用扣子
AI办公助手,复杂任务高效处理。办公效率低?扣子空间AI助手支持播客生成、PPT制作、网页开发及报告写作,覆盖科研、商业、舆情等领域的专家Agent 7x24小时响应,生活工作无缝切换,提升50%效率!


堆友
多风格AI绘画神器
堆友平台由阿里巴巴设计团队创建,作为一款AI驱动的设计工具,专为设计师提供一站式增长服务。功能覆盖海量3D素材、AI绘画、实时渲染以及专业抠图,显著提升设计品质和效率。平台不仅提供工具,还是一个促进创意交流和个人发展的空间,界面友好,适合所有级别的设计师和创意工作者。


码上飞
零代码AI应用开发平台
零代码AI应用开发平台,用户只需一句话简单描述需求,AI能自动生成小程序、APP或H5网页应用,无需编写代码。

Vora
免费创建高清无水印Sora视频
Vora是一个免费创建高清无水印Sora视频的AI工具
推荐工具精选





AI云服务特惠
懂AI专属折扣关注微信公众号
最新AI工具、AI资讯
独家AI资源、AI项目落地

微信扫一扫关注公众号